How bioluminescence can lead to next generation RTK drug discovery ?
Domain Therapeutics’ technology is based on genetically-encoded BRET (Bioluminescence Resonance Energy Transfer) biosensor assays for profiling tag-free cell surface receptor signaling and activity.
All of our technologies are created with the same underlying goal: Improve drug discovery by bringing new insights on transmembrane receptor’s proximal signaling transduction, without interfering with the structure of these receptors and effectors.
Both of these aspects are crucial for the development of next generation therapeutics based on a deeper knowledge
After having well validated the GPCR bioSens-All® platform, we transposed the same technology and its unique advantages to the realm of Receptor Tyrosine Kinases (RTKs). Given the importance of these receptors in the field of oncology, and the burden that these diseases represent, it becomes essential to bring new tools improving the drug discovery process in this field.
The importance of RTKs
RTKs bind to a variety of signalling molecules and regulate many critical processes, including cell survival, proliferation and differentiation. Importantly, dysregulated RTK function can affect many of the processes above and lead to the development of numerous types of cancer. Consequently, RTKs are prime targets for new anti-cancer agents.
Yet, our understanding of the complex biology of RTKs remains incomplete and, as a result, their full therapeutic potential is largely underexploited.
Current readouts limit the development of novel RTK therapeutics
Currently, it is well recognized that different ligands binding the same receptor can promote different biological outcomes, a concept known as functional selectivity (or biased signaling). Functional selectivity has been extensively described for GPCRs, yet this novel pharmacological paradigm remains underexplored for RTKs in part due to an emphasis on the discovery of ATP-competitive tyrosine kinase domain inhibitors. Biased ligands hold therapeutic promise due to their ability to selectively modulate beneficial signaling cascades while lacking activity on those that may produce adverse side effects.
Additional efforts and technologies are therefore required to fully understand the proximal and distal mechanisms that underlie the functional selectivity of RTK ligands. Our understanding of the complexities underlying the activity and pharmacology of RTKs remains incomplete, thus hindering the development of new therapeutics targeting specific downstream pathways.
Many of the current methods used to study RTK activation and screen for RTK inhibitors revolve around their kinase activity. However, kinase activity– based assays are limited in throughput and overlook additional key determinants of ligand therapeutic efficacy, including kinetics, localization, and functional selectivity, leading to an incomplete view about a ligand’s signaling signature.
Such limitations require the development of novel tools to improve drug discovery in the field of RTKs.
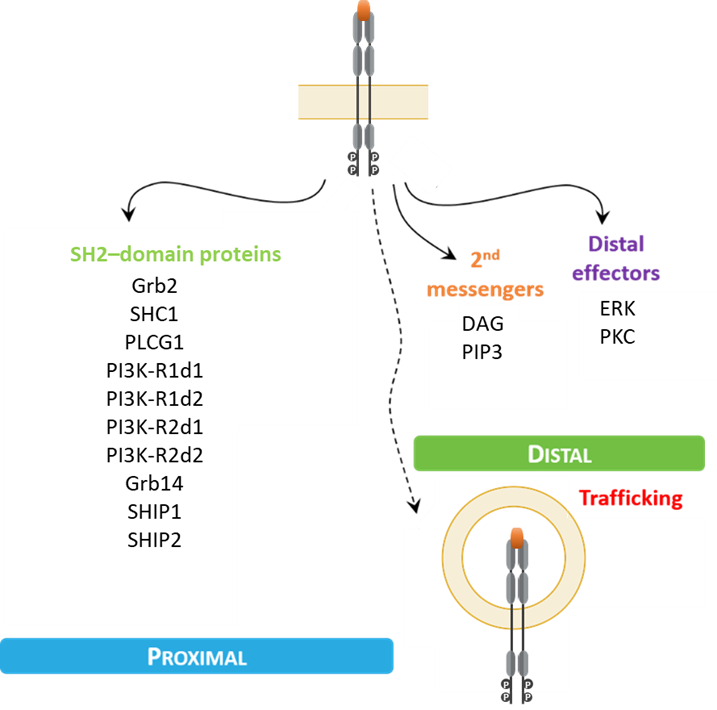
Signaling signature: A diverse set of SH2-domain biosensors for advanced ligand characterization
Upon activation by a given ligand (SME or biologic), RTKs go into dimerization process, which activates the autophosphorylation process, allowing the recruitment of SH2 domain effectors at the phosphorylated sites. The recruitment of these SH2 effectors represents the beginning of the intracellular signal transduction, determining the upcoming biological response of the cell.
Hence having the capacity to determine which SH2 effectors are recruited, and at which extent, can help further determining what biological response your drug could create. Given that every ligand can recruit these effectors at different levels, they all have their own signal transduction profile, that we call “signaling signature“. Knowing this information right from the beginning of a drug discovery project (hit / lead optimization stages) can be a game changer, improving the therapeutic effects and lowering the probability of failure of a given program.
To date, we have developed biosensors that monitor the activation of 12 distinct SH2 domain–containing proteins (Grb2, SHC1, PLCG1, PI3KR1-d1, PI3KR1-d2, PI3KR2-d1, PI3KR2-d2, Grb14, SHIP1, SHIP2, SHP-1, and SHP-2). The proximal biosensors forming
the basis of the RTK biosensor platform contain a specific SH2 domain of the previously named proteins fused to Renilla luciferase (RlucII). The recruitment of biosensors to the plasma membrane upon RTK activation translates into an increased BRET efficiency with a plasma membrane–anchored Renilla reniformis rGFP (rGFP).
Additionally, the same translocation principle is used to measure the internalization of RTKs with an early endosome–anchored rGFP.
A valuable information proven to be translatable to in vivo physiology
This proximal signaling information would not be so useful if we were not able to correlate it to in vivo effect and biology. Thankfully, since the signals we detect are very proximal to the plasma membrane, this information is physiologically relevant, meaning that we can observe correlations between therapeutic / adverse effects and a ligand’s signaling signature. Hence, having this information right from the early development of a drug helps you to take increase your chances of selecting / designing the right compound, with optimal therapeutic effects, and minimized attrition risks.
Tag-free RTKs for unbiased and reliable signaling data
A very distinctive and important aspect of our technology relies on the fact that neither the receptor nor its direct effectors are modified. Leaving this system in a physiological way guaranties that the signaling pathways activated are not altered by any structural change.
We do know for sure that a single amino-acid modification can highly modify a receptor’s biology, sometimes from partial to total loss of function. The beauty of our BRET technology also relies on its simplicity, we know that what we detect is physiologically meaningful.
Real-time kinetics: Additional insights for improved drug discovery
All of our SH2-domain biosensors can be monitored in real-time kinetics, providing additional insights on RTKs biology and pharmacology. Ultimately, this layer of data can be useful to discriminate ligands displaying similar endpoint signaling signature while having similar physiological effects,
This type of readout is still underexploited in typical drug discovery programs, making this feature interesting to develop the next generation of RTK drugs.
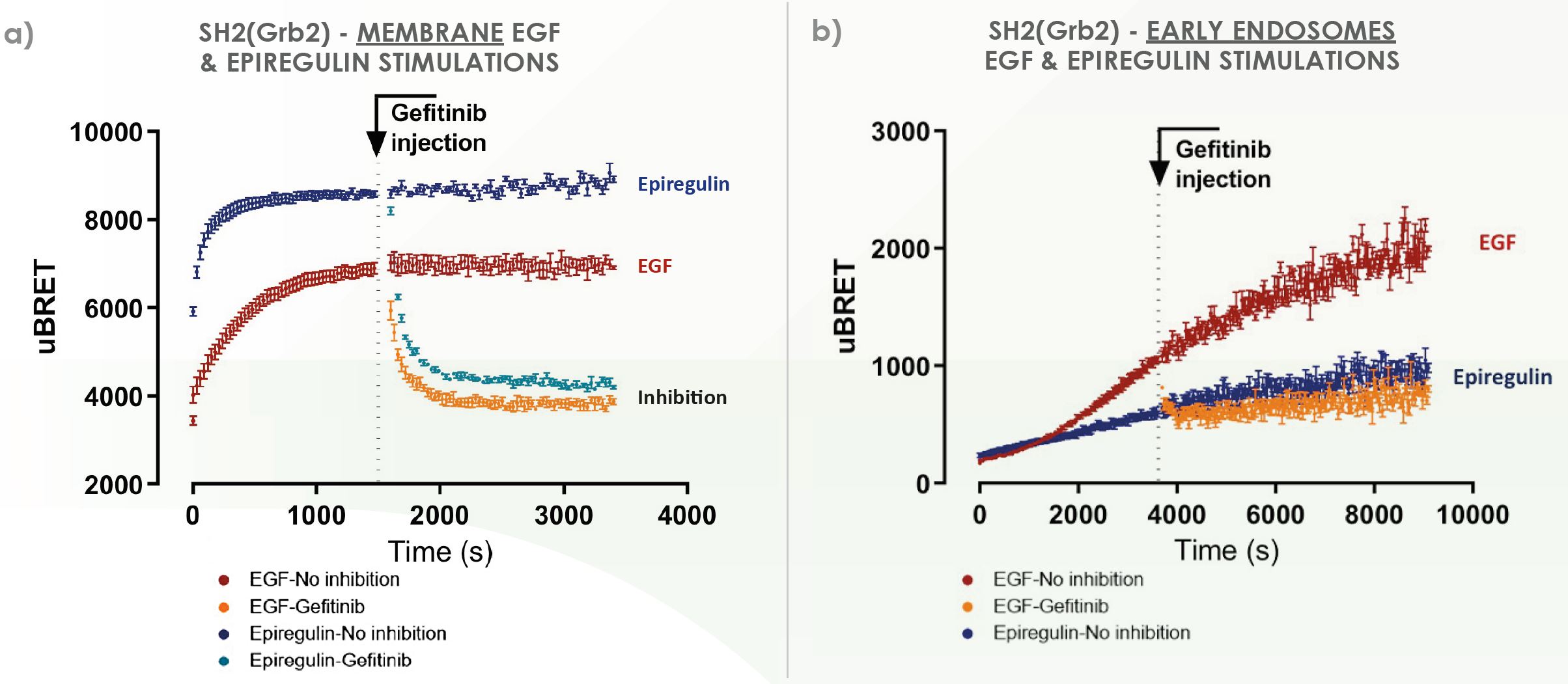
See here our case study exploiting real-time kinetics data.
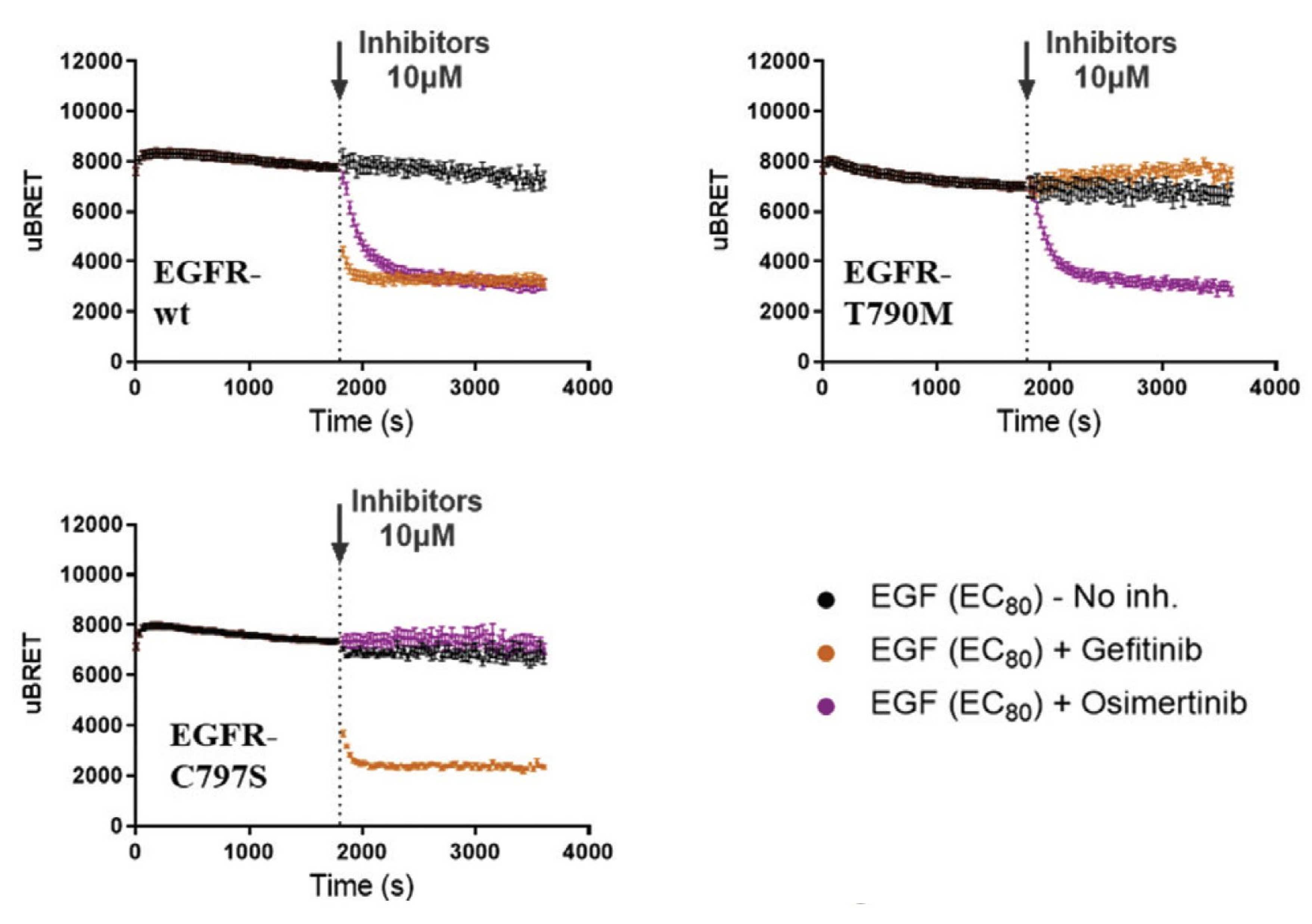
Mutant characterization for the development of next-generation medicines
The efficacy of several current drugs is limited by the development of drug-induced adverse events. Furthermore, the emergence of somatic and acquired RTK mutations allow tumors to develop resistance to certain drugs. RTK mutations may alter receptor activity and/or prevent drug binding through various mechanisms (that is, alteration of receptor subcellular localization, dimerization, signaling/trafficking, and kinetics signaling bias). The RTK biosensor platform allows us to differentiate the impact of different TKIs on various RTK mutations. This feature can be exploited as a tool to develop next-generation TKIs effective against RTK variants.